the Ultimate Deconvolution Experience
a true story
In May 2008 a letter arrived with the subject “iNMR suggestions”:
My name is Craig Robertson, a Ph.D student from the University of St Andrews. I work for Douglas Philp and he suggested I should get in contact to offer some suggestions which I feel would improve some of your software.
As you may guess I do a lot of NMR experiments, typically observing a reaction by 1H or 19F NMR for 16 h by recording a spectrum every 15/30 minutes. At the end of the day I have between 30-50 NMRs to deconvolute and typically look at 7-10 signals in each. To say it is tiresome is an understatement! Within our group we have different preferences on how to do this, I like using 1D WinNMR, (rather old) and others in my group use Topspin (A program I personally do not really get on with). We all use iNMR everyday for all our NMR needs except deconvolution, but with your introduction of the tool a little while back, we have had a look into it. There are a couple of things I would love to see before I can dump 1D WinNmr (And windows PC's altogether!) .
[note: Deconvolution had been introduced in October 2006 and Craig was probably the first customer to write about it, almost 2 years later.
The letter went on with several suggestions which mostly demonstrated that Craig had never found the manual page describing the deconvolution module, something he soon admitted. That's curious, because that module sports a question mark icon labelled “Help”.
It's also revealing: it looks like we stubbornly keep writing boring manuals that no user is ever going to read. Not to mention atypical articles like this one.]
The letter finished with the following wish:
The holy grail for a deconvolution tool would be one which could watch a peak evolve over many spectra. ie a sort of automatic deconvolution tool. I don't know if this sort of thing is possible however I thought I might mention it just in case. : )
At the time of the above email, iNMR was already able to perform almost everything that Craig needed.
The trick was as simple as copy & paste.
It was indeed possible to copy the whole list of parameters from the deconvolution module,
edit it into any text-editor (for example: adding or deleting lines) and pasting the list back
into the same or a different deconvolution module. Yes, they are non-modal windows, you can create how many of them you like
and they can even survive detached from their respective experimental spectrum.
What wasn't possible yet was the “automatic deconvolution” tool, but it didn't take long...
There were two problems: the module had never been tested by the public (the so called beta testing).
The solution, in this case, is simply to keep working on the details. The big problem was, as the reader has already realized,
that Craig had thousands of peak to process! iNMR already had the technology to handle the situation,
because it's a scriptable program, yet the deconvolution module had never been connected with the built-in Lua interpreter.
There were also other chemical and spectroscopic issues, but they are outside the scope of iNMR.
If you want to argue if deconvolution is really better than trivial integration,
then our answer is that, in this particular case, it is. Here is a representation of the first experiment
we received from Craig. For clearness, only the peaks increasing during the reaction are shown,
(those decreasing are important as well). You can download the spectra in
iNMR format from here if you want to recreate the ultimate deconvolution experience for yourself...
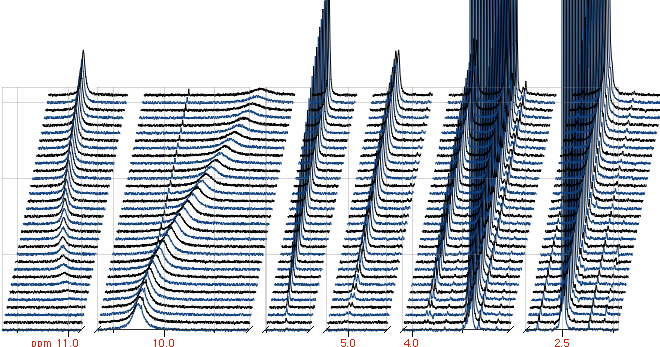
After a single working day we had already created a communication door between the Lua interpreter and the deconvolution module,
which had become fully scriptable in this way. The same day our
first script was also ready to run.
The whole “work” required to process the desired 160 signals consisted into the pushing of a single button.
The results were saved in tabular form as a text file:
point ppm area ppm area ppm area ppm area ppm area
01 3.710 158.981 3.927 1.120 5.665 0.000 5.008 10.222 3.775 6.660
02 3.710 156.606 3.926 1.294 5.641 2.275 5.008 1.785 3.725 17.781
03 3.710 154.335 3.925 1.133 5.665 2.194 5.017 2.212 3.735 15.102
04 3.710 152.229 3.924 2.634 5.669 3.756 5.014 3.490 3.749 10.079
05 3.711 147.785 3.919 8.495 5.671 5.686 5.011 5.136 3.753 12.886
06 3.711 144.402 3.920 9.880 5.672 7.793 5.009 7.181 3.755 17.001
07 3.711 139.776 3.918 13.994 5.673 10.168 5.007 9.103 3.757 20.275
08 3.711 135.007 3.919 16.619 5.674 12.742 5.006 11.519 3.758 24.961
09 3.711 129.215 3.918 20.249 5.674 15.181 5.005 13.960 3.759 29.479
10 3.711 124.476 3.918 24.391 5.675 18.184 5.004 16.737 3.759 34.418
11 3.711 118.477 3.918 26.400 5.675 20.961 5.003 19.377 3.760 40.019
12 3.711 113.019 3.917 31.753 5.675 23.793 5.002 21.912 3.760 46.052
13 3.711 107.711 3.917 35.634 5.675 26.979 5.001 24.823 3.760 51.704
14 3.711 101.458 3.917 38.670 5.675 29.406 5.000 27.455 3.760 56.673
15 3.711 96.096 3.916 41.935 5.675 32.424 5.000 29.934 3.760 61.986
16 3.711 90.981 3.916 46.517 5.675 35.176 4.999 32.905 3.760 67.574
17 3.711 85.938 3.916 50.616 5.675 37.724 4.998 35.648 3.760 72.676
18 3.711 80.728 3.916 52.905 5.675 39.995 4.998 37.794 3.759 77.344
19 3.711 75.670 3.916 56.149 5.675 42.612 4.997 40.455 3.759 82.661
20 3.711 70.732 3.915 58.468 5.675 44.740 4.997 42.319 3.759 86.801
21 3.711 66.532 3.915 61.093 5.674 47.021 4.996 44.713 3.759 90.900
22 3.711 62.266 3.915 63.992 5.674 49.090 4.996 46.455 3.759 95.325
23 3.711 58.436 3.915 66.910 5.674 51.181 4.995 48.559 3.758 99.654
24 3.711 54.914 3.915 68.394 5.674 53.240 4.995 50.574 3.758 103.199
25 3.711 51.620 3.915 71.398 5.674 55.086 4.994 52.366 3.758 107.380
26 3.711 48.574 3.914 72.650 5.674 56.493 4.994 53.913 3.758 110.269
27 3.711 45.332 3.914 74.438 5.674 57.942 4.994 55.334 3.758 113.179
28 3.711 42.629 3.914 75.678 5.673 59.555 4.994 56.913 3.757 115.742
29 3.711 40.732 3.914 77.763 5.673 61.135 4.993 58.331 3.757 118.953
30 3.711 37.836 3.914 79.841 5.673 62.104 4.993 59.270 3.757 121.244
31 3.711 35.633 3.914 81.420 5.673 63.317 4.993 60.662 3.757 123.958
32 3.712 33.316 3.914 81.753 5.673 64.431 4.993 61.836 3.757 125.846
are the numbers meaningful?
The table above has been compiled from 160 different deconvolution modules,
each one employing a (potentially) different unit to measure areas.
The unit for frequencies is reported (ppm) but not the unit for the areas.
How can we compare the 160 values? All we can do is to compare the values on the same row (same 1-D spectrum).
From these values we can calculate the percentage of each compound at different times.
Only the percentage values (not shown) can be used to monitor the kinetic of a reaction.
The areas expressed by iNMR are normalized either internally or externally.
Normal integrals, those measured directly on the spectrum, can also be normalized
or not (in the latter case the normalization factor is 1). The normalization factor can change from page to page.
Each deconvolution module, when created, inherits the normalization factor in use at the moment,
but only if it's different from 1. In the latter case, the modules are normalized internally, instead,
in order to make their total area = 100. If we want to compare the areas evaluated by different modules,
as in the case described, we must ensure ourselves that the normalization factor is the same
for the peaks on the same row. It doesn't mind, instead, if it changes from spectrum to spectrum,
because we can (and should) compare the variations in percentage and not variations in area
among the spectra. In conclusion, we need not to compare areas between different spectra, but we that:
- each spectrum is normalized before the first deconvolution
- the normalization factor is different from 1
- the normalization factor remains the same as long as we remain on the same row
Our 1-D spectra are extracted from a 2D matrix just before using them
and a new page is created by the extraction mechanism. The normalization factor of any new page is 1,
exactly the value we must avoid. It is necessary, therefore, to normalize the integrals
before starting the first deconvolution.
All iNMR users know how to pick the integrator tool,
double click an integral and enter the desired value into the dialog that appears.
The command “intreg()” can perform the equivalent operation inside a script (macro).
We must be sure, of course, that an integral already exists. Another way to normalize the integrals
is to create the first integral, because it's automatically normalized.
This alternative trick assumes that no integral exists. Our program adopts the latter alternative
and, to be sure that there are no integrals, we explicitly delete them:
delint() -- this command deletes all integrals
region( 2.4, 2.6 ) -- any region internal to the spectrum is good
press 'i' -- creates the first integral, automatically normalized
-- we have finished doing the necessary things
-- now we add something unnecessary:
intreg( 1, 300 ) -- we set our integral = 300
The unnecessary last line of the script adopts a meaningful scale for the areas.
We have intentionally selected a region comprising the methyl signals of both the starting material and the product,
and set its value = 300. In this way, any integral, divided by the number of underlying nuclei, already
corresponds to the percentage concentration of the compound it belongs to, IF THERE ARE NO ERRORS.
This particular normalization simplifies the visual inspection of the table of results, but it's not correct
to assume that the integrals correspond to the percentages.
If the methyl signals move (as they slightly do)
or if an impurity appears during the reaction, the calculated values don't correspond
to the percentages anymore.
how can we trust this script?
It's immediately evident that the value 10.222 (third from the right in the first row of our table) is wrong.
It should be near to zero, because it measures the product of the reaction and the first spectrum
contains almost no product. It is necessary to repeat the deconvolution manually or to
ignore this value. In our opinion, at the early stages of the reaction, when the product is
almost invisible, only the methyl singlets can be used to monitor the increase in concentration.
Another prudential rule,
at all advancement stages, is to avoid signals with severe overlap, if more resolved signals are available.
That is to say that it's not necessary to always use the same method to estimate the percentage concentrations.
A problem still remains: how do we know what's happening during the script?
Can we be sure that the optimal operations are performed each time?
The solution is to modify the script and allow the user to suspend the run, change the parameters and the operations,
to select with the mouse the regions that are fed into the deconvolution modules and to restart the run.
We have actually split the original script into two new ones:
An initialization script that is called only once
and a step forward script that is called twice for each signal.
After the initialization, the program shows the first peak. The user selects the portion
of the spectrum to process, then clicks on the button associated to the step forward script.
The second script creates the deconvolution module and fills it with the appropriate parameters, then exits.
Now the program shows the deconvolution window with the tentative parameters
for the peaks. The user clicks the “FIT” button. If he likes the fit,
he clicks the step forward button again.
This time the script stores the values, closes the module, then jumps to the next peak.
The cycle continues until all the peaks of all the spectra have been processed.
The scripts take care of the book-keeping:
extracting all the 1-D spectra from the 2-D matrix, zooming into the regions of interest,
summing up the individual intensities to obtain the area of each multiplet,
storing the results into a file. The user acts as a supervisor, while all the boring bureaucratic
activities are delegated to the scripts. Each agent (the user and the computer)
takes care of what it's more good at.
While the scripts are only valid for this specific spectrum, they can be easily adapted
to many similar cases. It's enough to redefine the parameters. They are conveniently
listed (and commented) at the beginning of the initialization script.
The only other statement that may require a change is the optional normalization we have already seen:
intreg( 1, 300 )
but it can also be left there without harm.
What's important, in all cases, is that you don't measure the kinetic directly from the areas,
but from the percentage concentrations derived from those areas.
conclusion
The deconvolution module is based on a graphic interface. Built over this basis, a purely textual approach
is also available. On top of both approaches, everything can be programmed by a skilled user, in full or in part.
If he can't write the script, like in the case of Craig, we do it for free.
It's even possible to add more buttons (inside the console) to adapt the interface
to the individual needs and tastes.
In this article we have seen how a long-standing problem was elegantly solved in a few working days.
For a more in depth analysis the curious reader is invited to study the lua scripts (see links above).
More introductory articles on the same subjects are:
acknowledgments
We thank Craig Robertson for the initial input, the helpful discussions and his testing activity.
We also thank Prof. Douglas Philp for letting us publish their spectra here.
