About Spin Systems
iNMR simulates 1-D spectra using a special type of document. When saved to disk, these documents use the extension .spins. The commands to create and change these documents are “New Spectrum” and “Define Systems”, respectively, under the menu Simulate. This page explains the options offered by the dialog that appears in both cases.
Quick creation
If you already have a formatted list of peaks like this:
5.250 (1H, dd, J=7.993, J=2.057), 5.356 (1H, dd, J=14.856, J=2.057), 6.739 (1H, dd, J=14.856, J=7.993)
you can conveniently bypass the definition of the systems. Just copy the list (for example: the text above),
go to iNMR and press ⌘-N (on the Mac) or Ctrl-N (on Windows).
This quick method does not work if two Js are identical. In this case, use the general method.
If you only know the formula, you can generate the required list by using the free NMR Predictor.
You can create one or many systems
A single document can host many separate systems. Separate means that no coupling exists between nuclei belonging to different systems. Two systems can correspond either to different molecules or to moieties of the same molecule. This sort of factorization not only requires less memory and less computing time, it also simplifies the management of parameters. To create (delete) a system, click the square button with the plus (minus) sign. You move from system to system with the “system no.” menu You can define from 1 to 30 systems. Each one has its own table of parameters and can have its own value of population (the equivalent of concentration, in arbitrary units). The absolute value of a population has no significance. Only the population ratio matters.
Table of parameters
Within each system, nuclei are labeled with capital letters: A, B, C... each in a different row. You can create more rows using the button “add row”. Another button removes the last row. In the case of magnetic equivalence, there can be two or more nuclei with the same label (e.g.: ABC3). You enter the number 3 at the intersection between the second column (labeled n) with the third row (labeled C). Before proceeding, make sure you understand the definition of magnetic equivalence. In the case of chemical equivalence (e.g.: AA'BB'), you cannot use the apostrophe and the system is redefined so that each nucleus has its own letter (the same example becomes: ABCD). In the case of chemical exchange, each system corresponds to an exchanging site. Nuclei with the same label (capital letter) can exchange. For example: you define three systems. If the first one is an ABC3 system, all other systems must also be ABC3. Nucleus B in the first system can exchange with Bs of other systems, but not with As or Cs.
Chemical shifts and chemical equivalence
You can optionally enter the chemical shift value, in ppm units, in the third column. Alternatively you can leave a zero and set the correct value later on, in the parameters list that appears at the side of the simulated spectrum. There are cases when you know that two nuclei have the same chemical shift. You have to tell it to iNMR, otherwise two unrelated parameters will be created. It is in your interest, indeed, not to define more parameters than those that exist in reality (the degrees of freedom of the system). You specify an equality using literal symbols instead of numbers. Take, for example, an AA'BB' system that you define as ABCD. You specify numerical values for two nuclei (let's say A=3 ppm and C=2 ppm). For B, instead of inserting a numerical value, you simply write “A” (without quotes). The chemical shift of D will be instead specified as “C”. From this moment on, iNMR will take care of the chemical equivalence. If you change the chemical shift of A, nucleus B will also move, and if you change the shift of C, nucleus D will move as well.
Symmetry
In the above paragraph we have seen an example of parameters entered in literal form. The principle is general and extends to coupling constants and to the parameters defined in previous systems. In the latter case, precede the parameter with the system number, for example: “1B”, “3A”, etc. Remember that you can only reference parameters defined in a previous system or in a previous row. You can also put a reference to a coupling constant defined in the same row, if it has been defined in a previous column. The reference to a coupling constant takes the form “JAB” or “1JAB”. The dipolar coupling takes the form “DAB”. The first letter (J or D) tells the kind of parameter, the second letter indicates the row, the third letter indicates the column. A line width can be referenced as “WA” or “W A” (with or without a space).
Scalar and dipolar coupling constants
For any two nuclei there are two points of intersection in the table. For example:
the row A intersects the column B and the row B intersects the column A.
This area of the table contains the coupling constants. For generic systems, only the upper triangle receives entries.
It means that you can specify JAB, but not JBA (the name of the row precedes the name of the column).
In this context “generic” means a spectrum in the liquid phase.
iNMR lets you also simulate spectra in liquid crystals, where residual dipolar couplings are visible too.
To activate this possibility, select “anisotropic”
in the radio group at the top-left and enter the dipolar coupling constants in the lower triangle.
It turns out that you can define DBA but not DAB.
Obviously, only one J and one D can exist for any given pair of nuclei.
While chemical shift parameters are always created by the program,
if you set a coupling constant equal to zero it is not created.
If, instead, you specify any non-zero value, you can modify it afterward, when the dialog is closed, inside the sidebar.
Symmetric systems with an even number of nuclei
Many symmetric systems can be divided in two halves, with no nucleus aligned along the symmetry element. You can easily recognize couples of nuclei with identical chemical shifts and identical coupling constants. Using the information here above (see the paragraph “symmetry”), it would be a good idea to describe one half of the molecule with numerical parameters and the other half with literal references. You have an even better solution. You select “symmetric” at the top-left. Now do not insert all the nuclei, but only half of the molecule. The number of nuclei corresponding to each letter is assumed to be 2 and therefore is omitted from the table. Insert the chemical shifts and couplings as in the generic case. Question: where will you put the Js between a nucleus in one half and another nucleus of the other half? Answer: along the diagonal or under it. Follow the example below and everything will become clear.
Example: ortho-dichlorobenzene
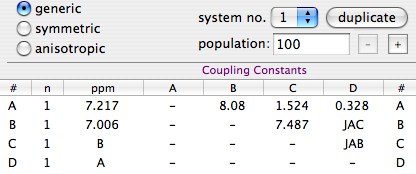
The solution in the first picture is aware of symmetry but not optimal. It explicitly declares only the minimal set of chemical shifts and couplings, but requires 4 rows and 6 columns. A, B, C and D correspond to nuclei H3, H4, H5 and H6, respectively, of ortho-dichlorobenzene. Note that the system is declared to be “generic”.
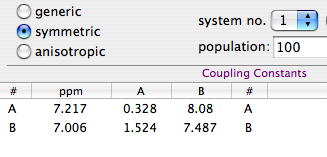
The second alternative requires only 2 rows and 4 columns.
It exploits all the advantages of symmetry (the system is declared to be “symmetric”).
The diagonal contains the couplings between nuclei with the same chemical shift. At the bottom-left,
the coupling JBA has been measured between nucleus A in one half of the molecule and nucleus B
in the opposite half (meta to each other). The value at the top-right has been measured inside
the same half (nuclei ortho to each other).
Note how the column “n” has been removed from the table.
X approximation
When the difference in chemical shift between two nuclei is orders of magnitude higher than their mutual coupling,
you are allowed to neglect non-diagonal terms in the Hamiltonian.
Peak intensities are in accordance with the binomial coefficients.
You declare the existence of one or more X nuclei and do not specify the chemical shift
(as it is treated as being at an infinite distance).
A typical case is the coupling between hydrogen and another nucleus like fluorine, phosphorus or carbon.
You can choose nuclei with any spin quantum number, but no more than a single set of them, all magnetically equivalent.
When the number of X nuclei is not zero, a new column appears in the table, whose header is “X”.
Insert therein the couplings with nucleus X.
There are cases in which a single X nucleus is not enough. For example, you are simulating a nucleus with two different hetero-nuclear couplings. You'll declare some hetero nuclei with the normal letters (B, C...). To tell iNMR not to display a given nucleus, set its chemical shift to a very large negative value (< -800 ppm).
Line widths
In the simplest case, all the lines in your simulated spectrum have the same width, governed by the parameter “defW”. To disable this option, you must open the usual dialog and uncheck it (you can read at the bottom of the dialog: “same width for all lines”). A new column appears in the table, where you can specify the individual widths, in Hz. If you do not specify them, the parameters are still created, all taking the default value “defW”.
Swapping labels
The button “Do” (swap) lets you exchange the position of any two rows in the table. The first column (#) is not affected, therefore the two nuclei not only swap their rows, they also swap their labels.
Related Topics
Parameters and Controls to Manipulate a Simulated Spectrum
Moving the Parameters into the J Manager